Clemens* AW, Wu* DY, Moore JR, Christian DL, Zhao G, Gabel HW. Jan 16, 2020, In: Molecular Cell. 77(2):279-293.e8. doi: 10.1016/j.molcel.2019.10.033. Epub 2019 Nov 26.
*Co-first authors
Abstract
The genomes of mammalian neurons contain uniquely high levels of non-CG DNA methylation that can be bound by the Rett syndrome protein, MeCP2, to regulate gene expression. How patterns of non-CG methylation are established in neurons and the mechanism by which this methylation works with MeCP2 to control gene expression is unclear. Here, we find that genes repressed by MeCP2 are often located within megabase-scale regions of high non-CG methylation that correspond with topologically associating domains of chromatin folding. MeCP2 represses enhancers found in these domains that are enriched for non-CG and CG methylation, with the strongest repression occurring for enhancers located within MeCP2-repressed genes. These alterations in enhancer activity provide a mechanism for how MeCP2 disruption in disease can lead to widespread changes in gene expression. Hence, we find that DNA topology can shape non-CG DNA methylation across the genome to dictate MeCP2-mediated enhancer regulation in the brain.
Department mini interview
Harrison Gabel’s laboratory studies neuroepigenomic mechanisms and their role in brain development and disease. We caught up with Dr. Gabel, Adam Clemens, and Dennis Wu to learn more about their recent study, MeCP2 represses enhancers through chromosome topology-associated DNA methylation.
DNA methylation is acknowledged as important for gene regulation in all cells. However, your work features studies of non-CG methylation in neurons, specifically. Why is this significant?
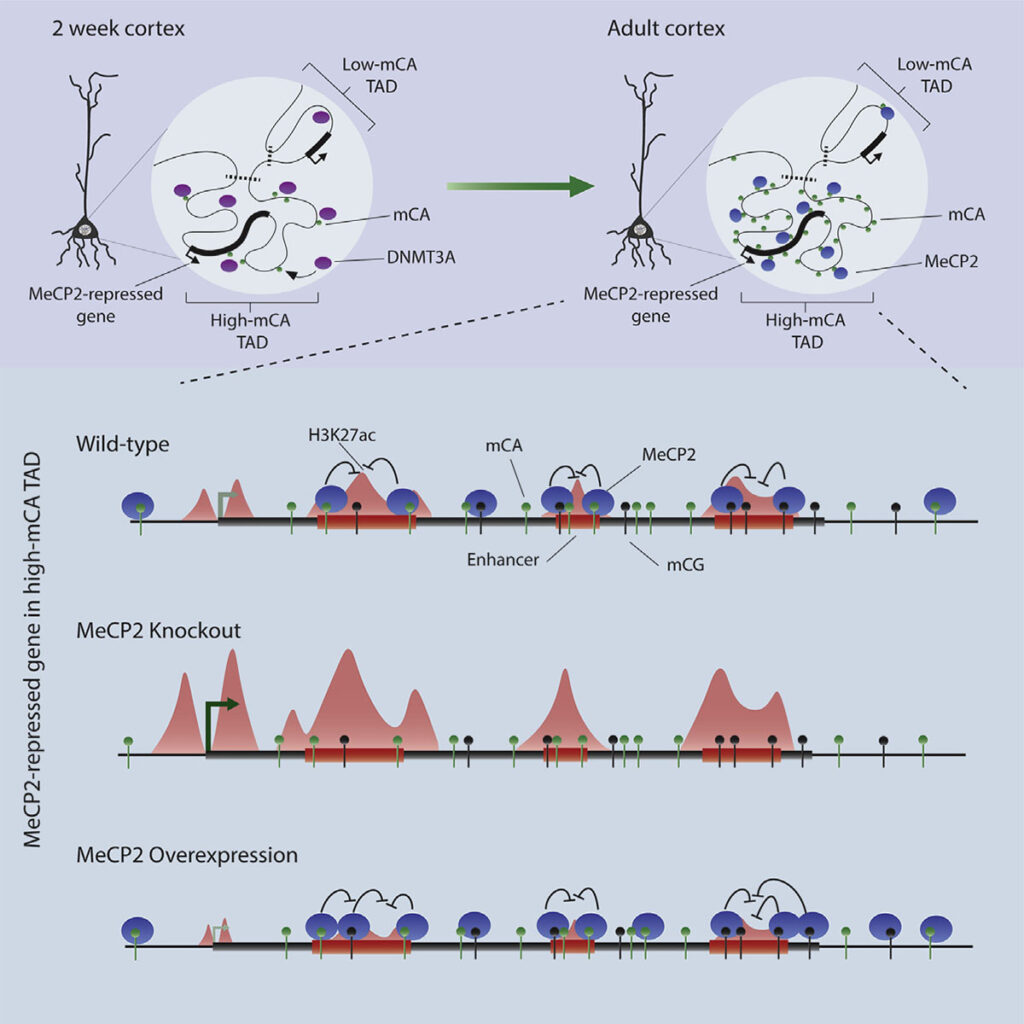
Adam Clemens: Methylation of cytosines in our DNA is a critical component of our epigenome, generally acting as a repressor of gene expression. Usually, this methylation is found at CG dinucleotides. However, a unique form of DNA methylation found at dinucleotides other than CG, non-CG methylation, is highly enriched within neurons of vertebrates. Why this non-CG methylation is needed in the brain, and how it contributes to gene regulation has been a bit of a mystery that we are trying to decode. Non-CG methylation accumulates in neurons in the early postnatal brain at the same time as the Rett syndrome protein, MeCP2. This suggests that these two neuron-specific elements could work together to regulate neuronal gene expression. Our lab previously showed that non-CG methylation is in fact a high-affinity binding site of MeCP2. In this study we started to decipher exactly how the MeCP2 and non-CG methylation collaborate to regulate gene expression.
In the course of the work, did you have an “aha” moment? Can you describe it?
Adam Clemens: The moment that sticks out to me is going to sound very simple, but I think it is actually an astounding feat of biology. It was previously shown that the neuronal genes most repressed by MeCP2 are long and coated in high levels of non-CG methylation, but the precise mechanism of repression for these genes was unknown. We first noticed that these same genes contain many regulatory sequences, called enhancers, and then obtained indications that MeCP2 act as a repressor by interacting with DNA methylation at those sites. This is when we suddenly realized that the repressive effects of MeCP2 at long and highly methylated genes could be explained by the presence of enhancers within these genes. This not only jump-started our progress in understanding gene regulation by MeCP2, but it also pointed to intragenic enhancers as uniquely important to control of transcription in neurons. This initial observation was a moment we knew we were onto something really exciting, with broad implications for the field of gene regulation.
Dennis Wu: For me, a key moment was when the sequence of the genome itself helped confirm our suspicions about enhancer regulation. Much of our analysis of the effects of MeCP2 across the genome depended on measurements of the binding sites for the protein. Biochemical studies have shown that MeCP2 prefers to bind methylated cytosine in CAC sequences much more than other trinucleotide contexts. After finding enhancers that we suspected might be directly regulated by MeCP2, we searched them for what sequences they contained. Strikingly, we found that methylated CAC sequences were specifically enriched in these enhancers. So not only was methylation enriched at these sequences, but the highest affinity site for MeCP2, methylated CAC, was present, insuring tight binding of the protein. This precise lock-and-key fit, jumped out to me as a strong support for functionally important binding of MeCP2 at these sites.
Your study touches on genome topology, making new findings about “topologically-associated domains”or TADs on chromosomes – how is this an important detail in the mechanism you are describing?
Dennis Wu: Genome topology, or the folding of the genome in the nucleus, is a relatively new area of research. Much is unknown about this process, but its importance is undeniable. TADs are defined as regions of the genome composed of hundreds of kilobases to megabases that clump and fold together in the same region of the nucleus, and they have been noted to impact a wide range of epigenetic marks. We found that TAD structures influence non-CG methylation patterns by guiding binding of the DNA methyltransferase 3A, or DNMT3A. Some TADs have high levels of DNMT3A binding during the early postnatal period and this results in high levels of non-CG methylation throughout the TAD. Other TADs are bound much less and have limited non-CG methylation. We found that patterning is very robust for neuronal non-CG methylation, setting it apart from typical CG methylation. Importantly, TAD-associated patterns of non-CG methylation help explain how the enhancers and genes most repressed by MeCP2 are found in megabase-sized regions that are enriched for non-CG methylation. Essentially, our thinking is that TADs dictate early developmental levels of DNA methylation, then when MeCP2 protein builds up, it can act in the large genomic regions enriched in methylation that were defined by the TAD structure.
You have mentioned the role of MeCP2, which, when absent or mutated, causes Rett Syndrome. MeCP2 is a very active focus of basic and clinical research – does your new model help frame the next set of questions for this important field, and how?
Harrison Gabel: Yes! In addition to providing some of the first mechanistic insights into how non-CG methylation can regulate genes, a major contribution of Adam and Dennis’s work is to give us a picture of what could be going wrong in the Rett syndrome brain. By pinpointing the regulation of enhancers as a critical site of action by MeCP2 that is disrupted in disease, we have updated the model for the molecular etiology of the disorder. This finding can motivate studies focused on enhancer dysregulation in the disorder, and perhaps inspire ideas for therapeutic development. In our ongoing work, have uncovered evidence that additional neurodevelopmental disorders related to Rett syndrome may arise, in part, from disruption of enhancer regulation by non-CG methylation and MeCP2. It is an exciting time to be exploring the unique neuronal epigenome and its role in brain function.